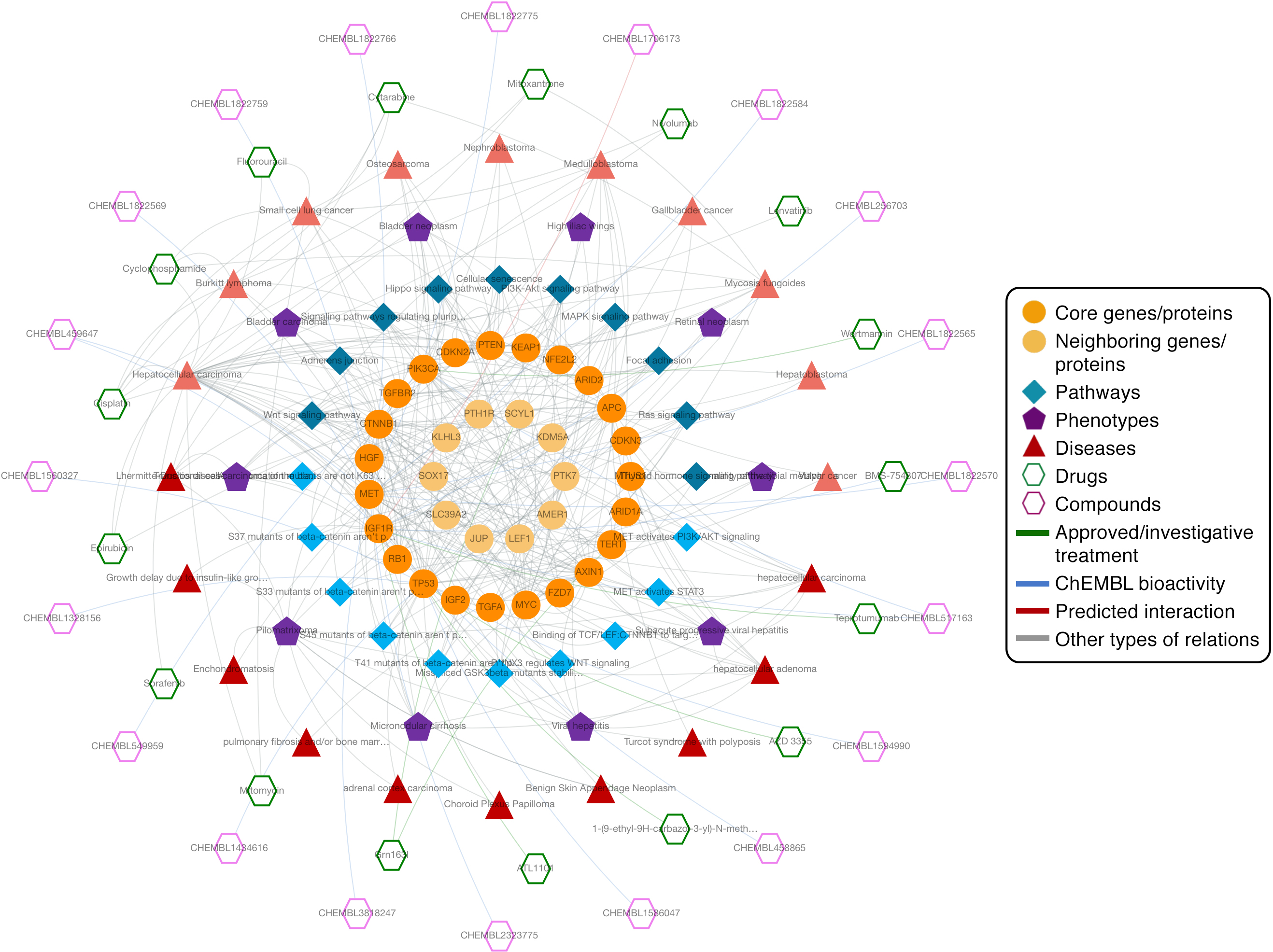
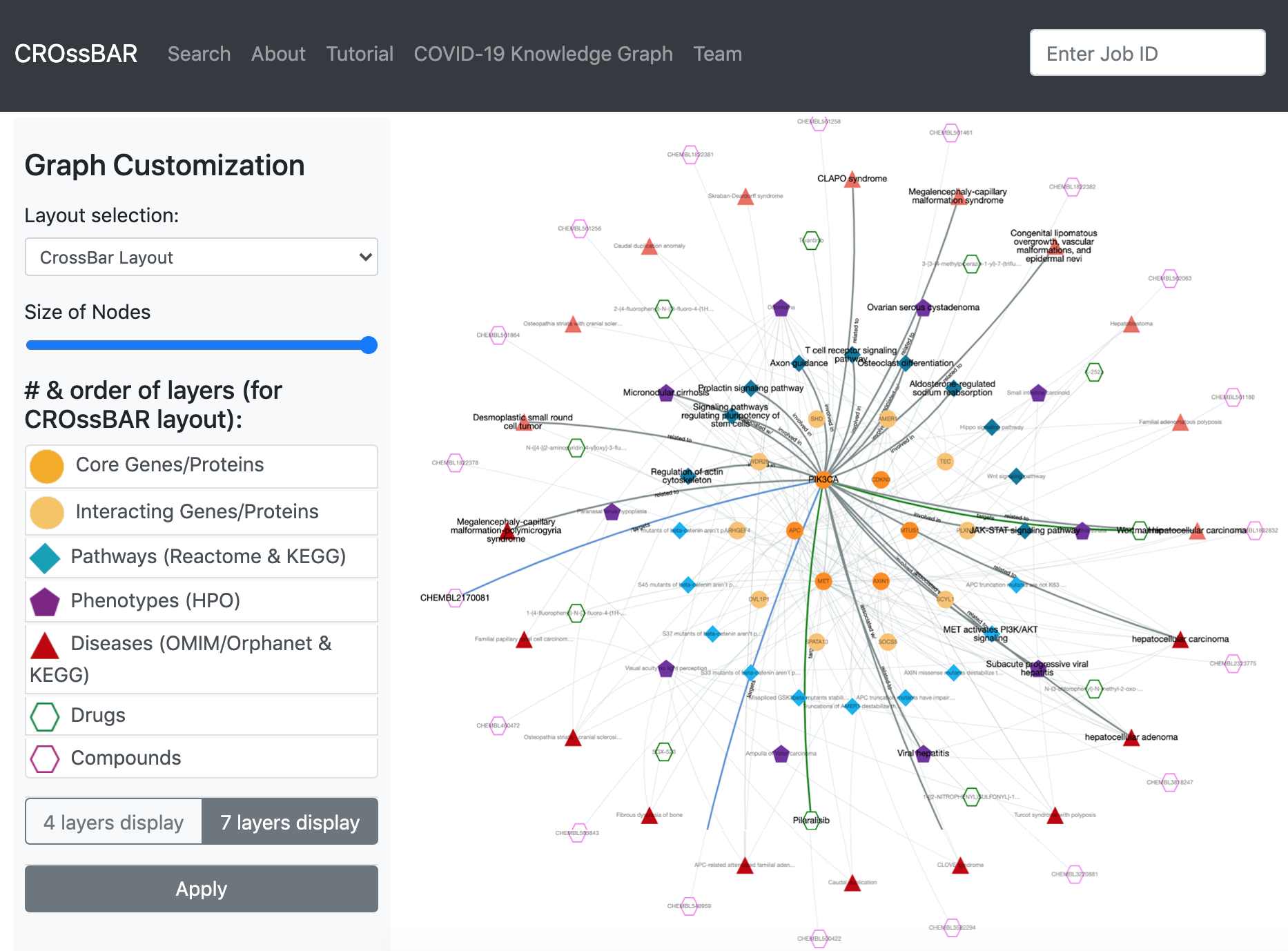
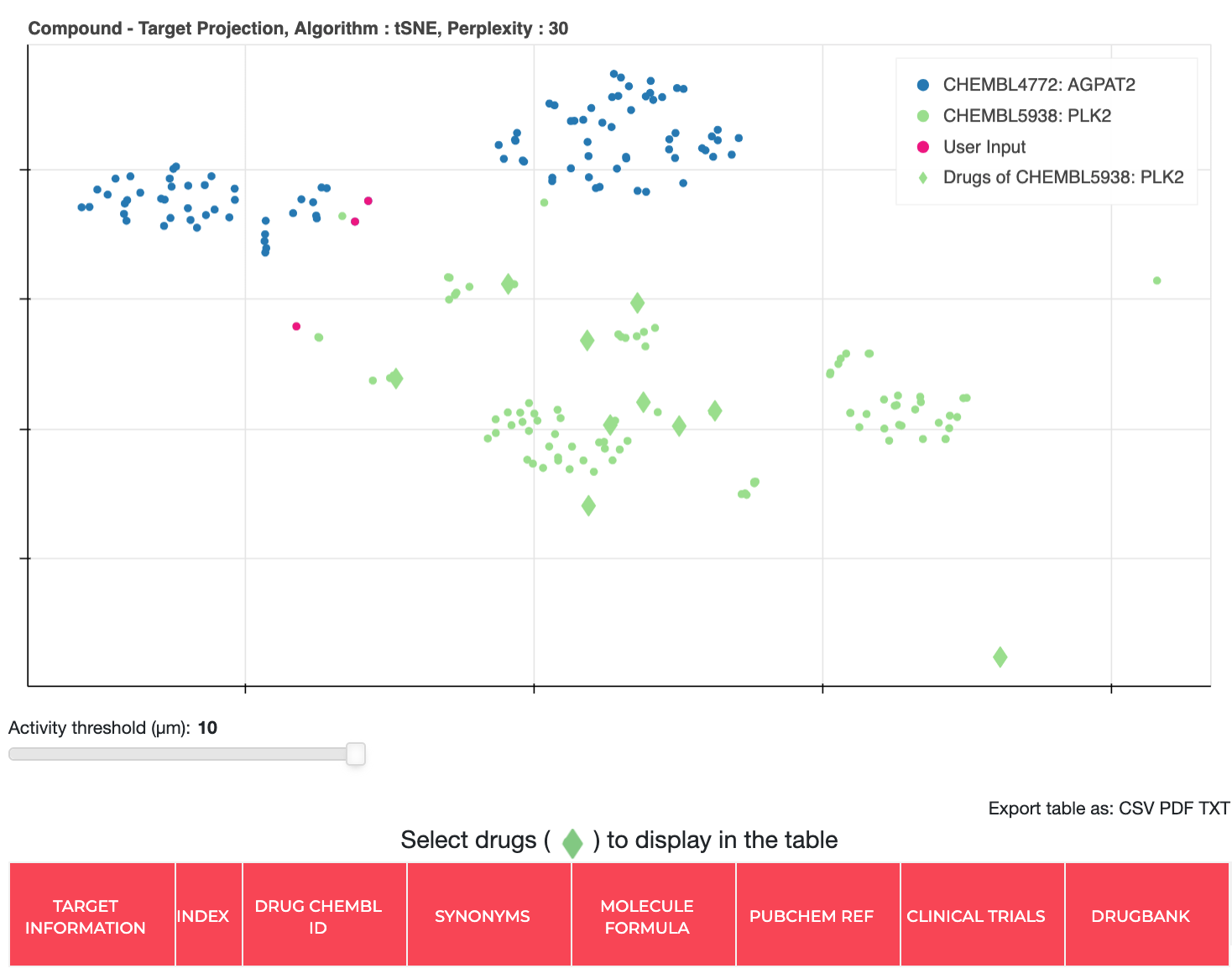
Other Work
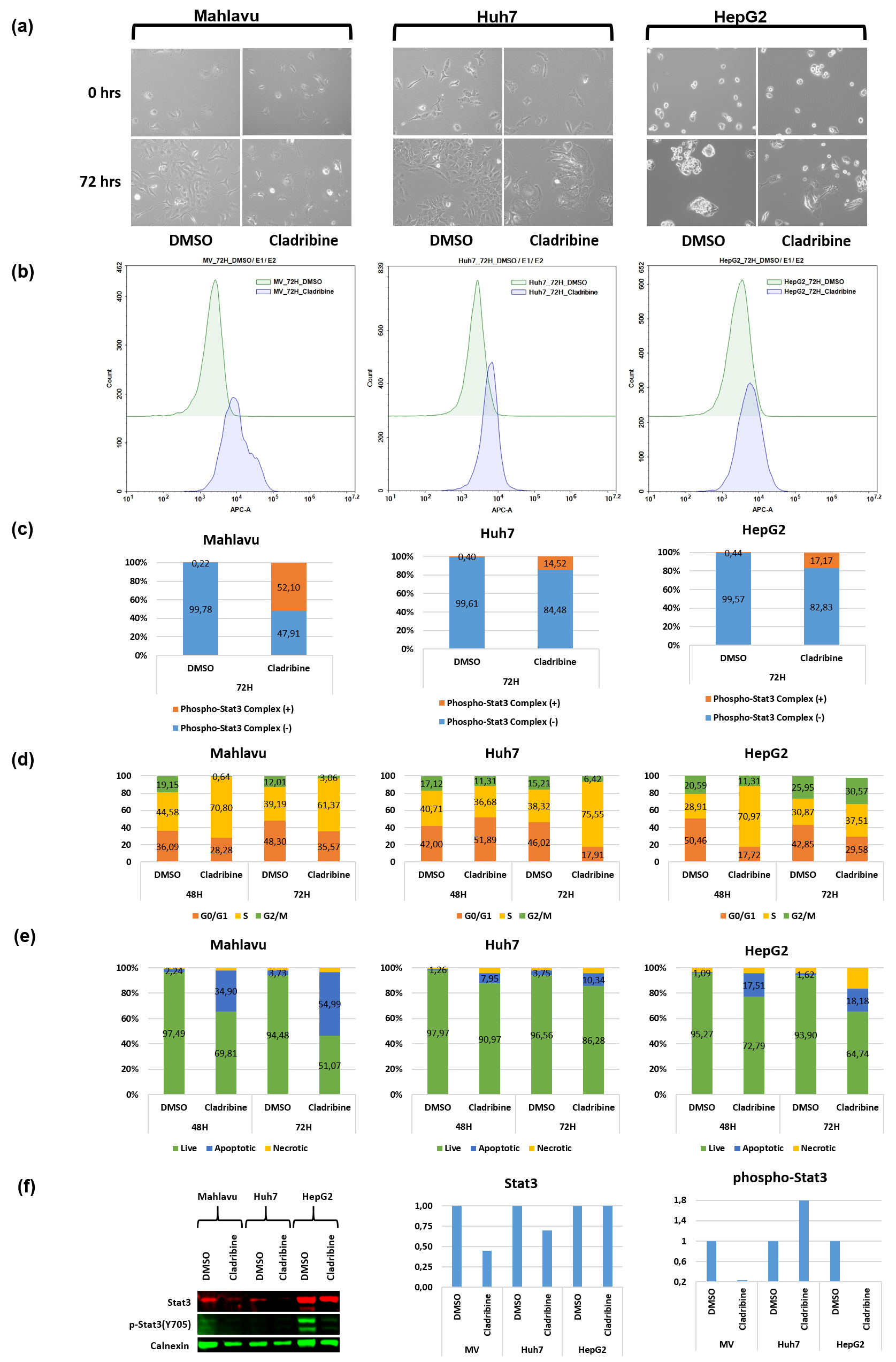
Abstract: Recent advances in computing power and machine learning empower functional annotation of protein sequences and their transcript variations. Here, we present an automated prediction system UniGOPred, for GO annotations and a database of GO term predictions for proteomes of several organisms in UniProt Knowledgebase (UniProtKB). UniGOPred provides function predictions for 514 molecular function (MF), 2909 biological process (BP), and 438 cellular component (CC) GO terms for each protein sequence. UniGOPred covers nearly the whole functionality spectrum in Gene Ontology system and it can predict both generic and specific GO terms. UniGOPred was run on CAFA2 challenge target protein sequences and it is categorized within the top 10 best performing methods for the molecular function category. In addition, the performance of UniGOPred is higher compared to the baseline BLAST classifier in all categories of GO. UniGOPred predictions are compared with UniProtKB/TrEMBL database annotations as well. Furthermore, the proposed tool's ability to predict negatively associated GO terms that defines the functions that a protein does not possess, is discussed. UniGOPred annotations were also validated by case studies on PTEN protein variants experimentally and on CHD8 protein variants with literature. UniGOPred protein functional annotation system is available as an open access tool at http://cansyl.metu.edu.tr/UniGOPred.html.
Publication : Rifaioglu, A.S., Doğan, T., Saraç, Ö.S., Ersahin, T., Saidi, R., Atalay, M.V., Martin, M.J., & Cetin-Atalay, R.,
Large-scale automated multi-functional annotation of protein sequences and an experimental case study validation on PTEN transcript variants Proteins. 2017;00:1–17. https://doi.org/10.1002/prot.25416
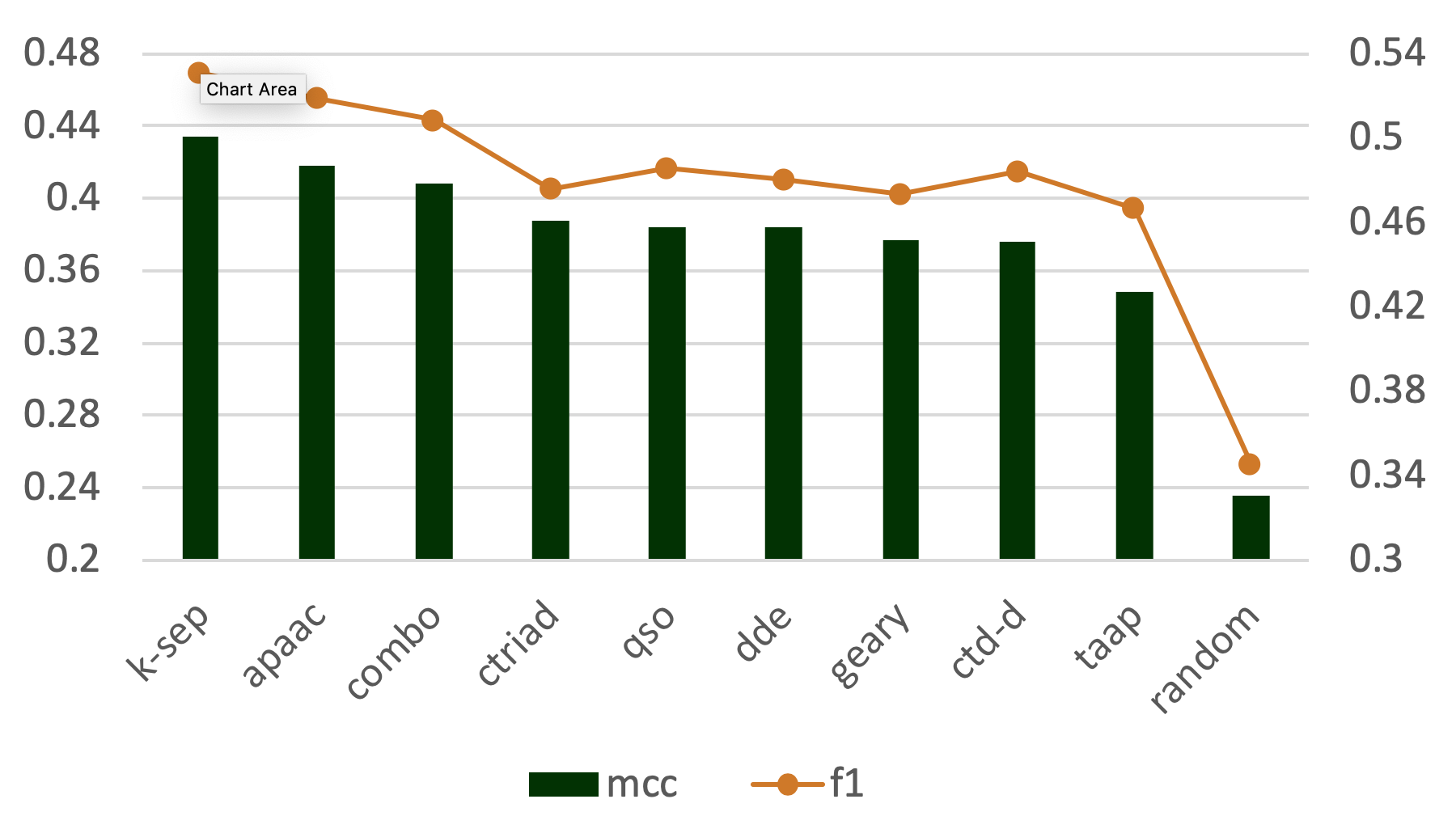
Abstract: The automated prediction of the enzymatic functions of uncharacterized proteins is a crucial topic in bioinformatics. Although several methods and tools have been proposed to classify enzymes, most of these studies are limited to specific functional classes and levels of the Enzyme Commission (EC) number hierarchy. Besides, most of the previous methods incorporated only a single input feature type, which limits the applicability to the wide functional space. Here, we proposed a novel enzymatic function prediction tool, ECPred, based on ensemble of machine learning classifiers. In ECPred, each EC number constituted an individual class and therefore, had an independent learning model. Enzyme vs. non-enzyme classification is incorporated into ECPred along with a hierarchical prediction approach exploiting the tree structure of the EC nomenclature. ECPred provides predictions for 858 EC numbers in total including 6 main classes, 55 subclass classes, 163 sub-subclass classes and 634 substrate classes. The proposed method is tested and compared with the state-of-the-art enzyme function prediction tools by using independent temporal hold-out and no-Pfam datasets constructed during this study.
Publication : Dalkiran, A., Rifaioglu, AS., Martin, MJ., Cetin-Atalay, R., Atalay, V. and Doğan, T.,
ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature ,
BMC Bioinformatics , 2018, 19:334,
https://doi.org/10.1186/s12859-018-2368-y
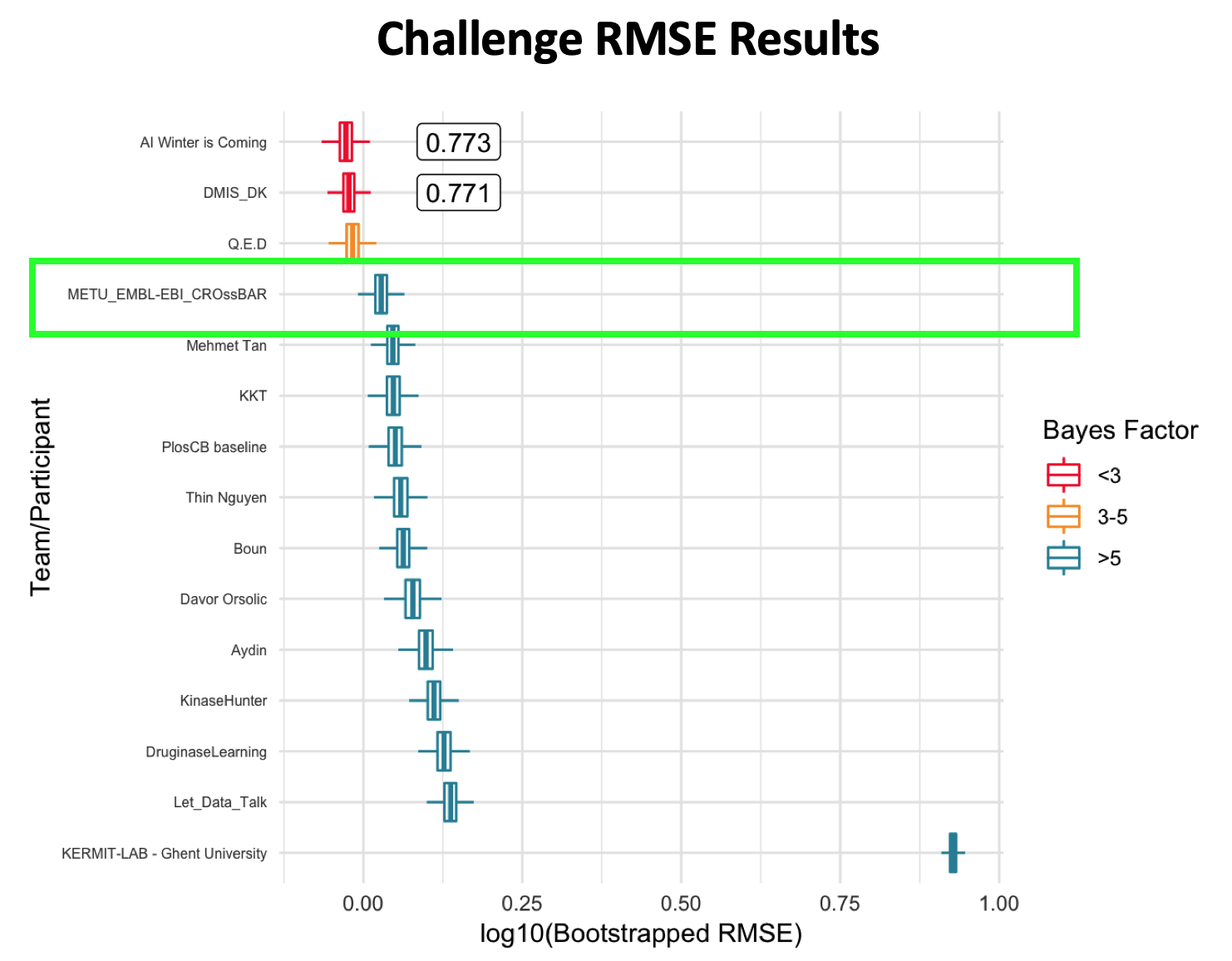
Abstract: Automated protein function prediction is critical for the annotation of uncharacterized protein sequences, where accurate prediction methods are still required. Recently, deep learning based methods have outperformed conventional algorithms in computer vision and natural language processing due to the prevention of overfitting and efficient training. Here, we propose DEEPred, a hierarchical stack of multi-task feed-forward deep neural networks, as a solution to Gene Ontology (GO) based protein function prediction. DEEPred was optimized through rigorous hyper-parameter tests, and benchmarked using three types of protein descriptors, training datasets with varying sizes and GO terms form different levels. Furthermore, in order to explore how training with larger but potentially noisy data would change the performance, electronically made GO annotations were also included in the training process. The overall predictive performance of DEEPred was assessed using CAFA2 and CAFA3 challenge datasets, in comparison with the state-of-the-art protein function prediction methods. Finally, we evaluated selected novel annotations produced by DEEPred with a literature-based case study considering the ‘biofilm formation process’ in Pseudomonas aeruginosa. This study reports that deep learning algorithms have significant potential in protein function prediction; particularly when the source data is large. The neural network architecture of DEEPred can also be applied to the prediction of the other types of ontological associations. The source code and all datasets used in this study are available at: https://github.com/cansyl/DEEPred.
Publication : Rifaioglu, A. S., Doğan, T., Martin, M. J., Cetin-Atalay, R., & Atalay, V. (2019).
DEEPred: automated protein function prediction with multi-task feed-forward deep neural networks ,
Scientific reports , 2019, 1 9(1), 1-16.,
https://doi.org/10.1186/s12859-018-2368-y